Manual
AlphaFind v2 is a search engine for protein similarity in the complete (214-million) AlphaFold (v4) [Varadi2022] and TED (365-million) [Lau2024] databases. It allows search at the protein-chain level, with residue-level pLDDT masking at fixed thresholds (70, 80, 90) to limit comparison to stable regions, and enables searches across the database of TED domains in a single-domain or multi-domain search mode that combines all domain matches within a single score.
The search works by fast approximate similarity search on vector embeddings of protein chains [Segura2025] and domains [Kandathil2025] followed by the search refinement using TM-Score [Zhang2022] on raw CIF files.
The search results are displayed along with metadata (name, organism), together with their TM-Score and RMSD measures (relative to the input protein). If the proteins are experimentally found, AlphaFind v2 also provides all associated PDB IDs.
Main page

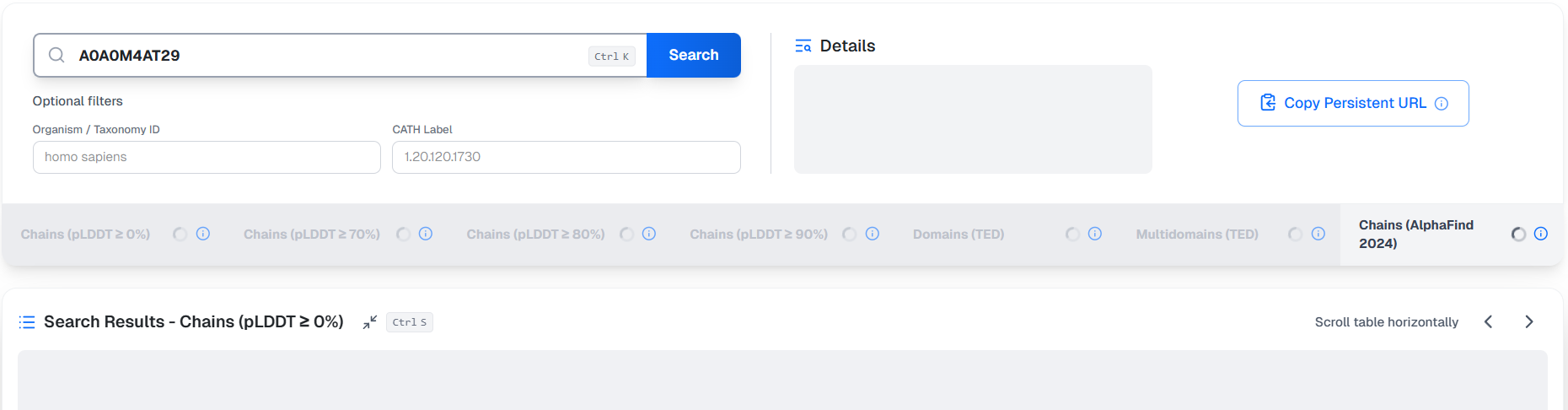
The main page features a search bar and three example use cases demonstrating the application's functionality. The search bar accepts various identifiers as inputs that are matched to the database contents. The searched database can be optionally constrained to a specific organism or CATH label. For example, it is possible to search for similar proteins only appearing in e.g., "Homo sapiens" or corresponding to a specific CATH label (e.g., "1.20.120.1730"). The CATH label is only applied to the domains search mode.
Clicking the Search button initiates all seven search modes that find structurally similar structures relative to the queried protein. The user is then redirected to the Results page.
Search input
The application accepts either UniProt ID (UniProt [UniProt2019] accession number), PDB ID (Protein Data Bank [PDB2000] accession number), Gene Symbol, or Protein's name.
Examples of accepted IDs by AlphaFind are: P13591 (UniProt ID) or P13591_TED01 (TED domain identifiers). The user can also input PDB ID (e.g. 12AS or "12AS:A". By default, chain ":A" is used when no chain is specified *), or a Gene Symbol (e.g. "HBA").
*AlphaFind is a similarity search tool for protein chains; therefore, searching with entire protein complexes is not possible.
Autocomplete
The autocomplete feature helps users resolve their search queries to match database contents in AlphaFind.
The resolution supports:
-
Protein names

-
UniProt IDs

-
Gene symbols

The user confirms their choices by clicking on the suggestion. The search with the selection is triggered by clicking on the Search button. The feature shows maximum of 5 suggestions at a time.
Optional filters
The search can be constrained to a specific cath annotation or organism (taxonomy). Organism is applicable to all search modes, while CATH annotation is only applicable to the domains search mode. Note that no filter is applied to the Chains (AlphaFind 2024) search mode.

In that case, the results shown will satisfy the filtering condition.
Note that the filters can be mutually exclusive, and as a result, no protein may satisfy the conditions and AlphaFind v2 will return empty results tables.
Results page

The Results page contains the input protein's metadata a link to dowload the raw mmCIF file and the number of combined search results. The user can also copy a persistent URL to the current search results to share with others or access later.
The search results are contained within seven tabs, corresponding to the seven search modes AlphaFind v2 uses. Each tab focuses on a different aspect of the input protein and contains different results.
Within each tab, the 3D View panel is shown on the left. On the right, the Search Results table is displayed. Each row of the table corresponds to a matched protein structure. The table is sorted by TM-Score alignment similarity ([Zhang2022]). If the TM-Score is not yet computed for a given row, the row is ordered according to the approximate embedding similarity ("kNN Score").
Progressive results loading
On search, the application will start fetching data from backend components. At first, no results are available:
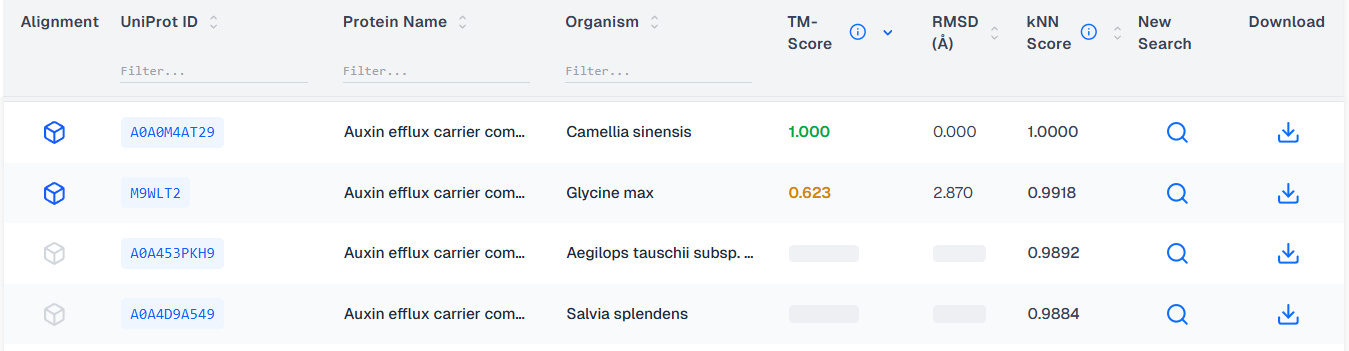
Usually, after a few seconds, the application will present results sorted by protein embedding similarity scores. At this point, superposition or TM-Scores are not available yet, but their computation has been triggered. The application iteratively fetches the computed superposition results and dynamically reorders the table by TM-Score, where available. This stage involves the most computation and can take some several seconds to minutes to fully complete, but the user can already interact with the results table.
Finally, the application finishes fetching the TM-Scores, the table's order is stabilized and all the alignments are ready. The next time the same protein is searched, the results will be present immediately.
Search Results table
The Search Results table for all chains search modes consists of these columns:
- Alignment: Button to trigger superposition alignment within the 3D View panel
- UniProt ID: UniProt identifier of the matched (target) protein.
- Protein Name: Name of the target protein.
- Organism: The organism in which the target was found.
- TM-Score: TM-Score as computed by USalign [Zhang2022] normalized by the input protein.
- RMSD (Å): Local measure of distance between the 3D coordinates of the aligned Cα atoms - the unaligned portions of the structures are disregarded.
- kNN Score: Embedding similarity, serving as a fast proxy for protein alignment.
- New Search: Button to trigger a new search with the target protein as the input protein. This search will open in a new tab.
- Download: Button for downloading the raw target protein structure as a mmCIF file.
The Search Results table for Domains (TED) search mode additionally contains:
- CATH Label: CATH classification of the matched (target) domain. This value can also be "-", in which case the domain is not classified by CATH.
The Search Results table for Multidomains (TED) search modes additionally contains:
- Domain pairs: List of domain pairs that are matched between the input protein and the target protein.
- Common domains: Number of domains that are common between the input protein and the target protein.
Additionally, the user can choose to extend the default columns with additional ones by clicking on "Show all" or :

- Target TM-Score: TM-Score as computed by USalign [Zhang2022] normalized by the target protein.
- Aligned residues: Portion of the aligned residues, relative to the total length of the input protein.
- Sequence identity: Portion of identical amino acid residues within the two aligned sequences, relative to the length of the input protein.
- pLDDT: Average pLDDT of the structure.
Experimental structures
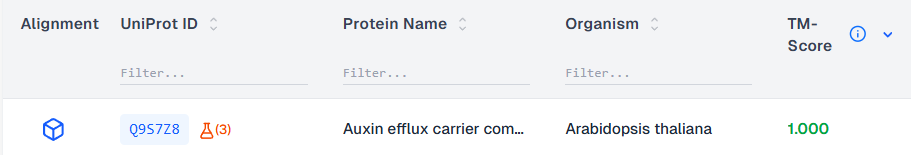
AlphaFind v2 also provides information about the experimental structures that the input or the target protein is associated with, if any. This information is displayed near the UniProt ID for the target:
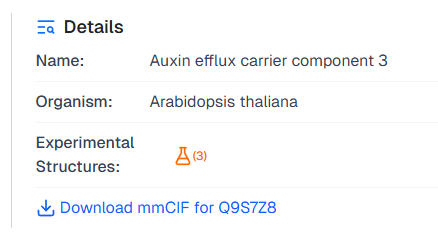
and in the Details panel for the input protein:
By clicking on the icon, AlphaFind v2 will open a dialog with the list of matched experimental structures:

Exporting results
The user can select the Export to CSV button at the bottom of the Search Results table to download the results as a CSV file.
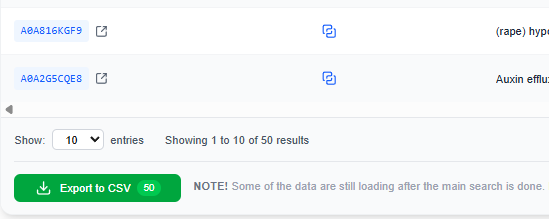
The CSV is semicolon-separated and contains the following columns for the chains indexes:
Organism;Protein Name;UniProt ID;TM-Score;RMSD;Aligned Residues (%);Sequence Identity (%);kNN Score;Experimental Structures;Gene;Tax ID
and
TED Domain ID;CATH Classification;TM-Score;RMSD;Aligned Residues (%);Sequence Identity (%);kNN Score;Experimental Structures;pLDDT;Organism
for the domains indexes.
Note that if the results in the table are not all present, the CSV will contain empty values for the missing values.
Loading more results
By default, AlphaFind v2 will load 30 results at a time for each search mode. To fetch more results, the user can click on the Load more button available on the last page of the results table:
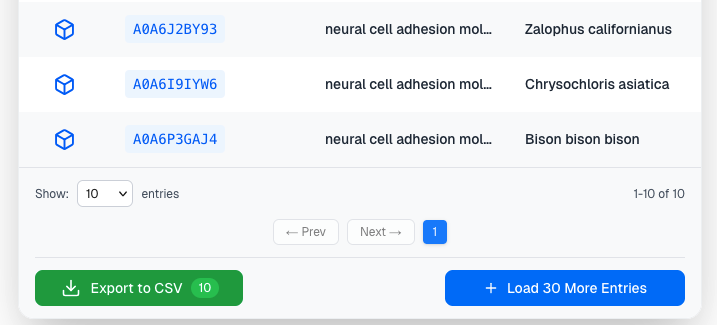
Load more button offers an option to select how many more new proteins will be loaded. On click, the application will fetch the next n results and add them to the table:

The table will be again reordered by TM-Score.
3D View
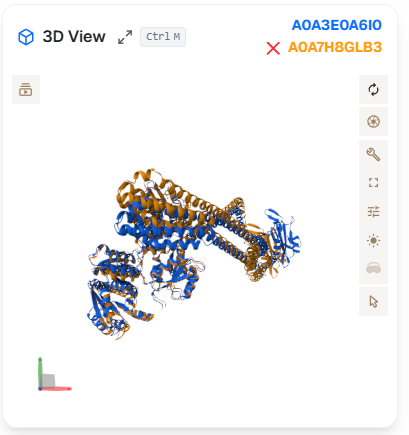
The 3D View panel uses Mol* [Sehnal2021] to visualize protein structures and their alignments. The user can rotate the structure by dragging with the left mouse button, zoom in and out with the scroll wheel, and pan the view with the middle mouse button.
The window can be expanded to cover 50% of the screen by clicking on the "Maximize" button.

In alignment, the 3D View shows the input protein in blue and the target protein in yellow. The alignment can be swiched off by clicking the red "X" button next to the UniProt ID of the proteins.
Tabs (Search modes)
AlphaFind v2 includes multiple complementary search modes to provide the user with different contexts for similarity, each suitable for a different use case. The tabs can be switched, the active tab is indicated by an underline. The numbers in the tabs indicate the number of results in the table.
Chains (pLDDT ≥ 0)
This search mode considers the entire protein chain from AlphaFold DB as is. It is suitable to use when the protein chain is stable or when its every part matters for the alignment.

Chains (pLDDT ≥ 90, ≥ 80, ≥ 70)
This search mode only considers residues of the protein chains that have pLDDT at least 90, 80, or 70. It is suitable for cases when the protein chain has low-pLDDT regions that are not important or would bias the structural alignment, and it is better to omit them.

In this case, the visualization shows the residues satisfying the pLDDT threshold in bright color; the remaining residues are shown in light blue.
This tab is only available if the input protein has a non-trivial portion of residues above the specified pLDDT threshold.
Domains (TED)
This search mode aggregates results for all domains found within the protein (if a whole protein chain was the input, e.g., "A0A3E0A6I0"). Additionally, it can be used to search for a single TED domain in case of a domain input (e.g., "A0A3E0A6I0_TED05").
The user can select among the available domains:

for each TED domain, the visualization highlights the identified domain within the protein:
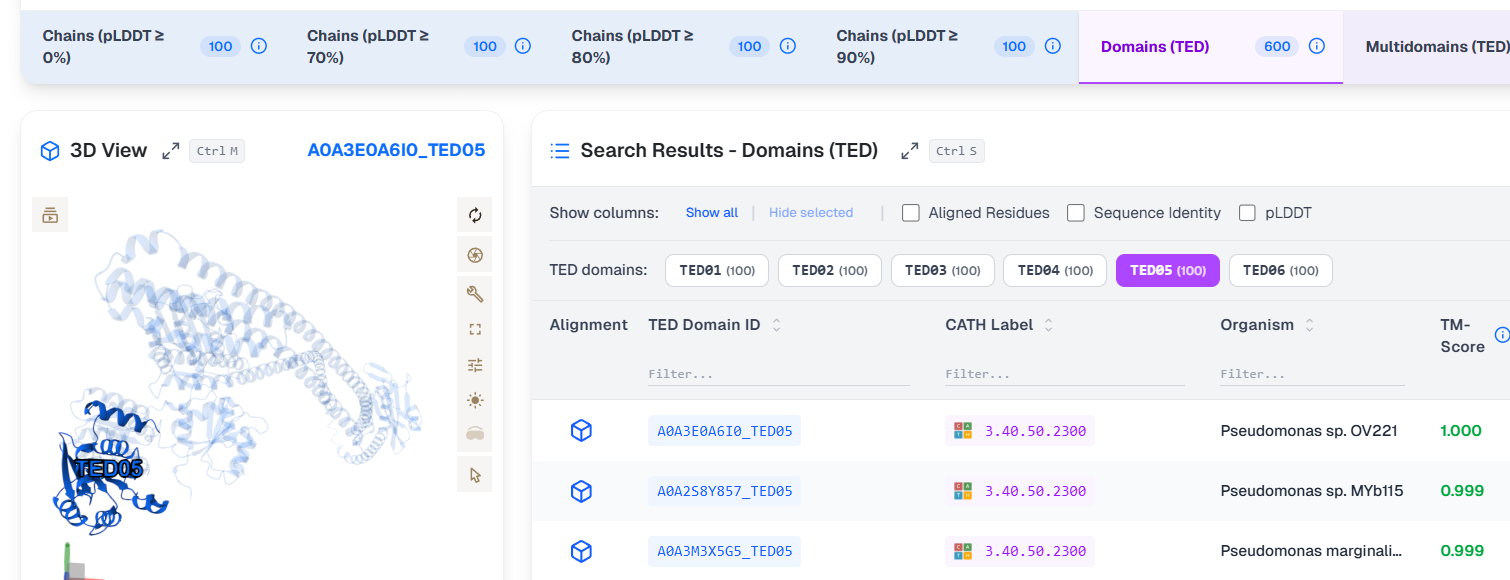
as well as within the target protein when superposition is selected:

This search mode is only available if the input protein has at least one TED domain.
Multidomains (TED)
This search mode integrates the search results from all the TED domains into a single table and reports the overall optimal superposition. Note that this search mode is only available if the input protein has at least two TED domains.

In the example above, the 3D View shows input protein's 7 TED domains.
In the Search Results, AlphaFind v2 reports on TM-Score, which combines all the domain alignments into a single score. On selection of the superposition, the 3D View shows the superposition optimized for the first domain match by default. The user can then use the Domain pair weights slider to adjust the weights given to the different domains.

For example, we can set the weights of all domain alignments to the same value, leading to a perfect superposition of the input protein's domains:

Details of TM-Score computation in Multidomain search mode
TED multidomain mode compares proteins as collections of independent domains.
The input protein is first split into its TED domains. Each input domain is searched against the database. This produces multiple matches to domains from many target proteins. A target protein may match the input in one domain or in several domains.
All domain matches are then grouped by target protein. For each target protein, we obtain a set of matched domain pairs: one input domain matched to one domain of that target protein. Which numbered TED domain matches which is irrelevant.
A multidomain score is computed from these matches. It is based on the TM-score formula but applied at the domain level. The score is the average over all input domains, not just the matched ones. Each matched domain contributes a value that depends on its RMSD: better structural matches contribute more.
Two scores are computed:
- A input-side score, normalized by the total number of domains in the input protein.
- A target-side score, normalized by the total number of domains in the target protein.
The score increases when more domains are matched and when the matches are structurally better. A perfect score of 1 occurs only when every domain in the protein has a matching counterpart with zero RMSD. Matching only a subset of domains lowers the score, even if those matches are perfect.
The relative positions of domains do not matter. Domains are treated independently, regardless of their order or spacing in the structure. Linkers, rearrangements, or unstructured regions have no effect. For this reason, the method is described as a “bag of domains” comparison.
Chains (AlphaFind 2024)
The final tab is the AlphaFind v1 application, as introduced in [Prochazka2024] and available at https://alphafind.fi.muni.cz/.

Limitations
- The application uses AlphaFold DB files in version 4, as the recent (October 2025) v6 update is not publicly available in its entirety.
- The results provided by AlphaFind are approximate; the engine can not guarantee that the best possible results for a given input protein will be found.
- The maximum number of results that can be fetched is 1000.
- The shortcut for maximizing “3D View” Command-M is used by macOS to hide the current window. Safari overrides the website's behavior by actually hiding the window, while other browsers (Firefox, Chrome) are not affected (the 3D View within the page is correctly maximized).
Bug Reporting & Feedback
If you encounter an error or have an idea for an improvement, please send a report to alphafind@ics.muni.cz. Thank you!
References
[Prochazka2024] Procházka, David, et al. "AlphaFind: discover structure similarity across the proteome in AlphaFold DB." Nucleic Acids Research 52.W1 (2024): W182-W186.
[Varadi2022] Varadi, Mihaly, et al. "AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models." Nucleic acids research 50.D1 (2022): D439-D444.
[Lau2024] Lau, Andy M., et al. "Exploring structural diversity across the protein universe with The Encyclopedia of Domains." Science 386.6721 (2024): eadq4946.
[Segura2025] Segura, Joan, et al. "Multi-scale structural similarity embedding search across entire proteomes." bioRxiv (2025).
[Kandathil2025] Kandathil, Shaun M., et al. "Foldclass and Merizo-search: scalable structural similarity search for single-and multi-domain proteins using geometric learning." Bioinformatics 41.5 (2025): btaf277.
[Zhang2022] Zhang, Chengxin, et al. "US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes." Nature methods 19.9 (2022): 1109-1115.
[Uniprot2019] UniProt Consortium. "UniProt: a worldwide hub of protein knowledge." Nucleic acids research 47.D1 (2019): D506-D515.
[PDB2000] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank (2000) Nucleic Acids Research 28: 235-242.
[Sehnal2021] Sehnal, David, et al. "Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures." Nucleic acids research 49.W1 (2021): W431-W437.